Cell News 3/2013
18
RESEARCH NEWS
dominant inherited forms characterized by severe to moderate
disproportionate dwarfism and pronounced joint laxity. In these
patients premature OA often requires early joint replacement.
The more severe form PSACH is caused exclusively by mutations
in COMP (Briggs et al., 1995; Hecht et al., 1995). Electron mi-
croscopy of PSACH patient biopsies showed that mutated COMP
is retained in typical granular or lamellar inclusions in the en-
doplasmic reticulum (ER) of chondrocytes (Maddox et al., 1997;
Stanescu et al., 1993). The accumulation of COMP in turn leads
to co-retention of other matrix components. Interestingly, col-
lagen IX and matrilin-3 are also retained but the trafficking and
secretion of collagen II remains mostly unaffected (Dinser et
al., 2002; Hecht et al., 2005). Impaired protein trafficking com-
promises normal ER function and eventually leads to ER stress,
which in turn results in an increased rate of apoptosis. Indeed,
in the growth plate of affected individuals many dead cells were
detected and a general disorganization with chondrocytes being
arranged in clusters rather than in columns was described (Ha-
shimoto et al., 2003; Hecht et al., 2004). It is therefore believed
that chondrocyte depletion in the PSACH growth plate is the
main reason for the diminished linear growth leading to dispro-
portionate short stature. The milder form MED has, in addition
to COMP, been linked to at least four other matrix genes coding
for each of the three chains of collagen IX and for matrilin-3
(Warman et al., 2011). Interestingly, mutations in matrilin-3
and the
α
3 chain of collagen IX lead to very similar intracellu-
lar inclusions as observed in PSACH patients (Bönnemann et al.,
2000; Cotterill et al., 2005). Based on the intracellular retention
of mutated proteins several studies have suggested that PSACH
and MED are mainly storage diseases of the ER. However, it was
demonstrated in cell culture models that not all disease causing
mutant proteins are retained. Instead, secreted mutant proteins
cause a disruption of extracellular matrix structures, indicating
that both intra- and extracellular pathogenic pathways can con-
tribute to the disease mechanism (Dinser et al., 2002; Schmitz
et al., 2006). Nevertheless, it is puzzling how mutations in genes
coding for structurally unrelated proteins cause a more or less
identical phenotype. Like COMP, matrilin-3 and collagen IX have
been shown to interact with each other to fulfill their structural
role in the ECM. Due to mutations in each of these proteins these
interactions might take place already intracellularly, leading to
premature formation of highly ordered but insoluble aggregates
(Merritt et al., 2007). A recent study has shown that retention of
mutant COMP in chondrocytes stimulates caspase-independent
necroptosis (Coustry et al., 2012).
Mouse models lacking cartilage matrix proteins
Collagen II deficient mice produce structurally abnormal car-
tilage and lack growth plates in long bones. As a result these
mice develop a skeleton without undergoing endochondral bone
formation. The phenotype is rather severe and these animals die
perinatally (Aszodi et al., 1998). Interestingly, collagen XI defi-
cient mice survive but develop features comparable to human
OA (Li et al., 1995). Mouse models deficient in the perifibrillar
proteins collagen IX, matrilin-3 and COMP have been generated
and analyzed intensively with regard to skeletal development.
Mice lacking collagen IX are viable and initially only minor ab-
normalities were observed, i.e. an osteoarthritis-like degenera-
tion in knee joints at later stages of maturation (Fässler et al.,
1994) and a retarded bone fracture healing (Opolka et al., 2007).
However, a more detailed characterization revealed that colla-
gen IX deficiency causes severe disruption of epiphyseal cartilage
architecture in newborn mice including a loss of the columnar
arrangement of chondrocytes in the developing growth plate
(Blumbach et al., 2008; Dreier et al., 2008). At the molecular
level, the deletion of collagen IX in mouse results in a reduced
integration of matrilin-3 into the cartilage extracellular matrix
and, to a lesser extent, of COMP (Budde et al., 2005). This confir-
med the notion that in vivo matrilin-3 is an interface component
interconnecting macromolecular networks and mediating inter-
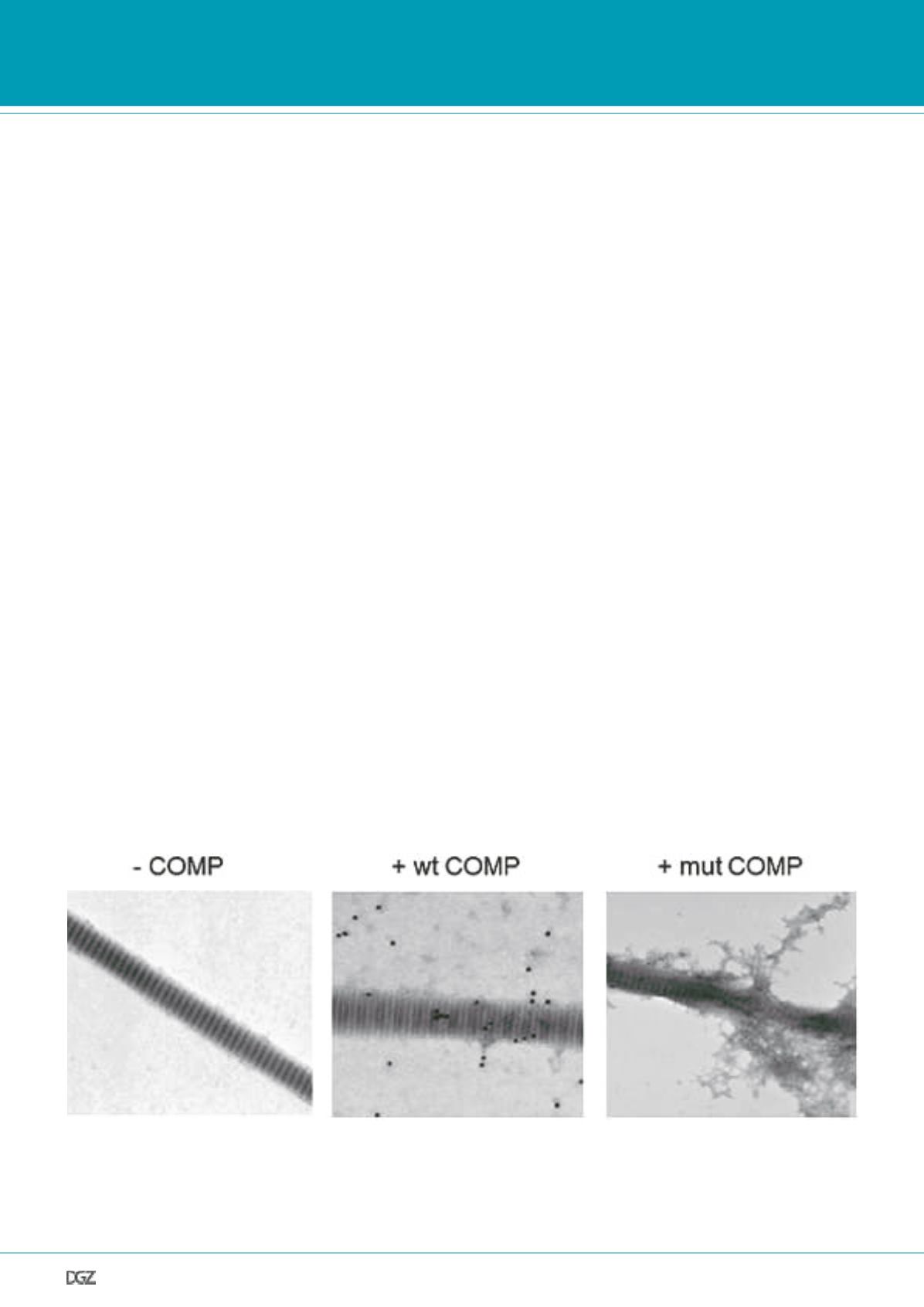
Figure 3. End products of collagen fibrillogenesis in the absence or presence of COMP.
In the absence of cartilage oligomeric matrix protein (-COMP), formation of typical banded type I collagen fibrils was observed (left panel), and in the
presence of wild-type COMP (+COMP), the type I collagen fibrils formed were identical to those in the absence of COMP (middle panel). Immunogoldla-
beling using a polyclonal antibody against COMP revealed labelling (black particles) on the formed fibrils as well as beside the fibrils. In the presence of
the mutant p.H587R COMP (+mut COMP, right panel), disorganized type I collagen fibrils with an irregular banding pattern were formed and amorphous
nonfibrillar material was often found.


