Cell News 2/2014
11
Research news
to missorting of Tau into dendrites, with subsequent decay of
microtubules. Consequently, mitochondria, vesicles, and other
cargo suffer from traffic jams in these dendrites. Neurofilaments
that are usually transported along microtubules from the soma
into the axon, become also mislocalized to the somatodendritic
compartment, which indicates impaired transport. Remarkably,
this loss of microtubules and synapses, aberrant mitochondria
and neurofilament distribution depends on Tau, as in neurons
derived from Tau knockout mice (TauKO) these pathological
changes do not occur (Zempel et al., 2013).
In search for the causes of microtubule breakdown in dendrites,
we found that the decay of microtubules is executed by spas-
tin, a microtubule severing enzyme. Spastin in turn is recruited
by polyglutamylation of microtubules, conferred by the enzyme
TTLL6 (Tubulin-Tyrosin-Ligase-Like-6), a microtubule modifying
ligase (Lacroix et al., 2010). TTLL6 catalyzes the addition of po-
lyGlu residues within the C-terminal tail of
α
-tubulin, which in
turn leads to the recruitment of the microtubule severing pro-
tein spastin and subsequent destruction of microtubules. TTLL6
is mislocalized from the soma into dendrites by a Tau depen-
dent mechanism, involving an interaction of TTLL6 with Tau’s
N-terminal half (Zempel et al., 2013).
Tau is a microtubule associated protein (MAP) capable of bin-
ding to and stabilizing microtubules. Proper axonal sorting of
Tau depends on intact Tau-microtubule interactions (Kanai
and Hirokawa, 1995). In the pathological condition of Tau mis-
sorting, however, Tau is phosphorylated at a number of sites,
including the KXGS-motifs in the repeat domain of Tau (Thies
and Mandelkow, 2007; Zempel et al., 2010). These sites regulate
the binding of Tau to microtubules and are targets of the Mi-
crotubule-Affinity-Regulating Kinase (MARK), which in mature
neurons is most active in growth cones and the somatodendritic
compartent, but not in axons (Timm et al., 2011). When phos-
phorylated at the KXGS-motifs, Tau cannot bind and stabilize
microtubules, and can become missorted (Li et al., 2011). Phos-
phorylation of Tau at the KXGS-motifs is essential for the loss of
microtubules, the loss of synapses mediated by Tau missorting,
the translocalization of Tau into spines, and destruction of spi-
nes and synapses (another pathological hallmark of AD). Con-
trary to earlier views, the majority of missorted Tau in the soma
and the dendrites does not originate by retrograde flow from the
axon, but from newly synthesized protein that is not properly
routed into the axon anymore (Zempel et al., 2013).
Significance and Implications for
Therapeutic Approaches
A key result is the identification of a novel reaction cascade im-
plicated in the missorting of the Tau protein in neurons affected
by AD. The results reveal a multi-step pathway leading from the
exposure of neurons to A
β
oligomers towards Tau-dependent
synaptic deficits. We identified new contributors and functions
in the pathological cascade of AD, centered around missorting
of Tau into the somatodendritic compartment. This includes the
decay of dendritic microtubules mediated by TTLL6 and spastin,
leading to Tau-dependent loss of spines in a phosphorylation
dependent manner. It provides an explanation for the well-
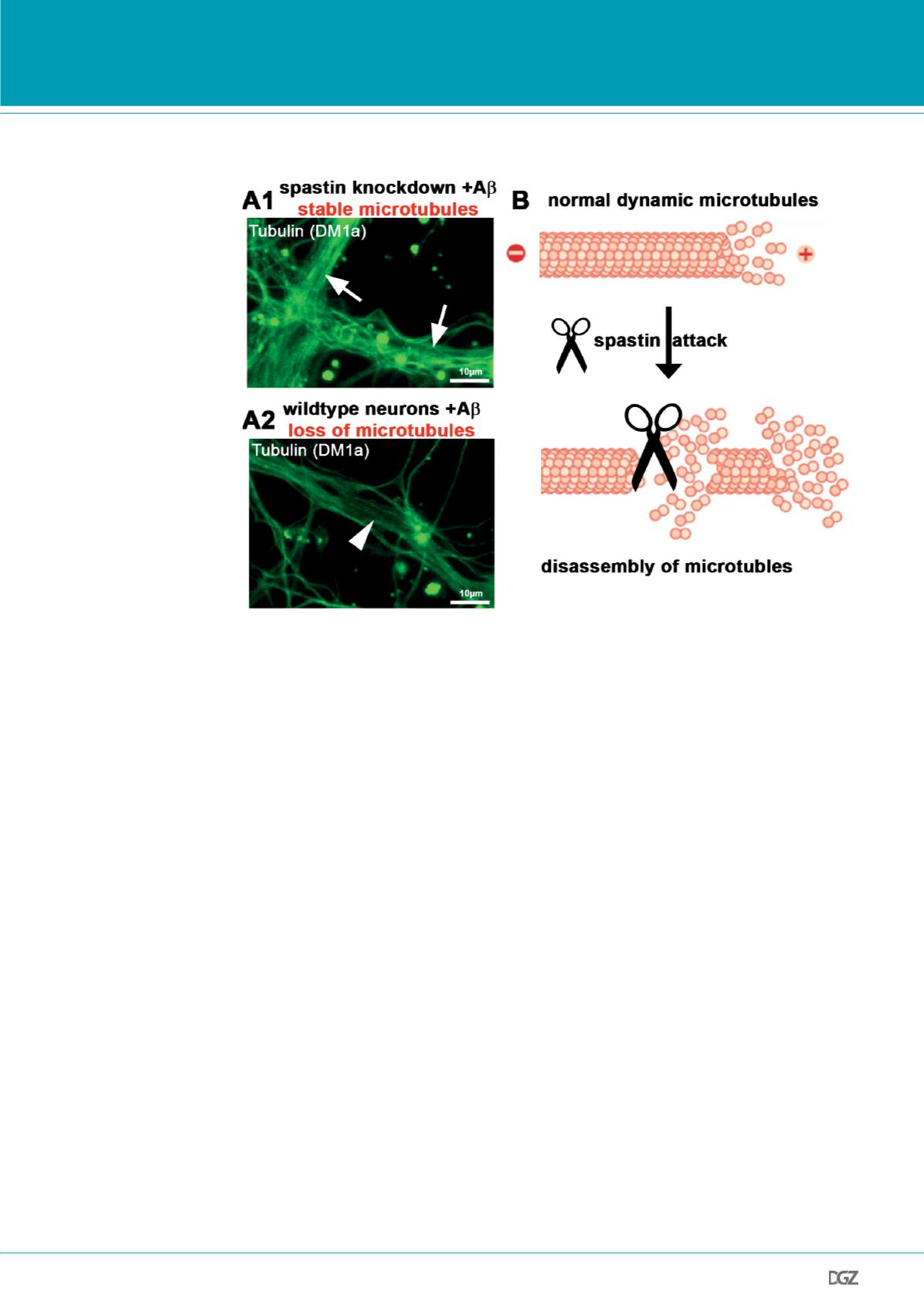
Figure 2. Spastin executes
microtubule loss after
invasion of dendrites by
Tau and TTLL6.
Spastin was silenced using
shRNA with a vector coex-
pressing RFP (5d) in primary
hippocampal neurons aged 16
days in vitro, cells were then
treated with 1μM A
β
for 3h.
A: Silencing of spastin results
in stable microtubules after
A
β
treatment. Cells expressing
shRNA (arrows, A1), show no
microtubule reduction. Neigh-
bouring untransfected cells
with normal spastin levels
show loss of microtubules in
dendrites after A
β
exposure
(A2, arrowheads).
B: Schematics of spastin medi-
ated microtubule disassembly
in dendrites in presence of
missorted Tau.
Modified from (Jean and Baas,
2013; Zempel et al., 2013).


